the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Jul 2018
| 11 Jul 2018
Towards the unravelling of the slug A. ater–A. rufus complex (Gastropoda Arionidae): new genetic approaches
María L. Peláez
Antonio G. Valdecasas
Daniel Martinez
Jose L. Horreo
The genus Arion includes several slug species, some of which are considered to be a pest to both cultivated and wild flora. Within this genus, the Arion ater complex comprises two different morphological forms: Arion rufus and A. ater, but there is no consensus about their species status. Their phylogenetic relationships have been recently solved, both of them belonging to different phylogenetic clades, but their species status is still unclear (as different clades are not always different species). For this reason, the aim of this study was to precisely identify these species status by employing the up-to-date multi-rate Poisson tree processes (mPTP) methodology as well as the classic methodology of genetic distances, both of which have three different mitochondrial genes. Results confirmed that both A. ater and A. rufus are independent evolutionary clades, and the high genetic distances between them (K2P distances ranged between 9.1 and 16.4 %, depending on genes) together with mPTP analyses, supported the idea that the clades correspond to different species. Results will be useful for the classification of these specific species as well as for developing proper pest control methodologies and conservation policies in both cultivated and wild plants.
- Article
(291 KB) - Full-text XML
- BibTeX
- EndNote





The genus Arion includes up to 50 slug species, several of which are of considerable agricultural and environmental concern (Barr et al., 2009). Some of the species in this genus are highly invasive, for example Arion vulgaris (Moquin-Tandon 1855), which is one of the worst invading species in Europe (Gismervik et al., 2014). Key in any strategy for pest control and conservation purposes is species identification (Amstrong and Ball, 2005), and in the genus Arion it is important to distinguish between closely related alien and native species. The correct identification of all Arion species must be a priority in order to properly face the diverse agricultural and conservation problems related to them but also to improve the taxonomy in the group.
Within the genus Arion, the Arion ater complex comprises two different morphological forms, Arion rufus and A. ater, but there is no general consensus about their species status (Hatteland et al., 2015). Some researchers consider them to be subspecies (e.g. Burnet, 1972), and others suggest that they could be species (e.g. Bank et al., 2007). In addition, there are some indications of hybrid forms (e.g. Hagnell et al., 2003) and introgression has occurred between them (e.g. Hatteland et al., 2015; Noble and Jones, 1996; Rowson et al., 2014). This problem probably comes from their very similar external morphology and their distinct internal morphology, as well as life history and ecological traits (Blattmann et al., 2013). For example, A. ater is in general black coloured and A. rufus is orange, while another species of the genus (A. vulgaris) is brown; thus specimens of dark brown can be misidentified (Roth et al., 2012), although the morphology of their genitalia separates clearly these species (Hatteland et al., 2015). Genetically, their phylogenetic relationships were initially not clear and they were even proposed to be ecotypes (Quinteiro et al., 2005), but more recently it has been suggested that A. ater and A. rufus are probably independent monophyletic clades (Rowson et al., 2014). But the fact remains that they belong to different phylogenetic clades and does not establish that they are actually different species. For this reason, the different genetic species identification methods that are currently available, and that are especially useful in these kinds of problematic cases (e.g. García-Jiménez et al., 2017), must be employed here.
Species identification techniques have been greatly improved in the last decades thanks to the increasing availability of genetic information (i.e. DNA taxonomy; Goldstein and De Salle, 2011; Vogler and Monagan, 2007). In fact, the use of DNA characters (e.g. genetic distances) could be useful in the formal naming of species (Renner, 2016), and it has been frequently employed for all kinds of organisms during recent decades. However, the barcode scheme (Hebert et al., 2003) is not usually enough for diagnosis, but the coalescent model is a new tool for testing whether the clades involved in the taxa under study are evolutionary independent (Zhang et al., 2014). In all cases, the main goal is to identify known species and delimit new ones, which can be rather difficult in many cases (Vogler and Monagan, 2007). The phylogenetic species concept (PSC) approaches the species delimitation problem, relying on the fact that phylogenetic species are the smallest units for which phylogenetic relationships can be reliably inferred from DNA data (Baum and Shaw, 1995). Inspired by this concept, the coalescent Poisson tree processes (PTP) model was developed in order to infer putative species boundaries (Zhang et al., 2013). This method is more robust and less affected by restricted sampling (Agrens et al., 2016), a main constraint limiting the use of other phylogenetic methods. It overcomes, too, the difficulty of having to deal, in many cases, with a single specimen per species. This situation is more common than usually acknowledged, as a great number of taxa have been discovered from a unique specimen, and may be called the singleton methodological dilemma (Lim et al., 2012).
For all these reasons, the aim of this work was to unravel the species identity of A. ater and A. rufus with different genetic techniques. This study included the recently developed PTP estimations as well as the classic genetic distances method, all of which were performed with the information contained, for the first time, in three different mitochondrial genes. It may confirm the validity of these potentially two different species, which again motivates the search of morphological correlates and may lead to improved identification in the field.
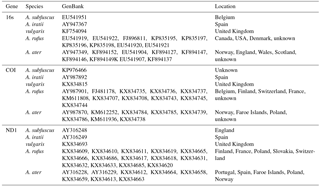
Because the phylogenetic information of a group of species can be contained in one or a few representative genes (Horreo, 2012), three mitochondrial genes were employed in this study (Table 1): 16s rRNA (16S), cytochrome oxidase subunit 1 (COI) and NADH dehydrogenase subunit 1 (ND1). They included sequences from different areas of Europe and America and their species identification was different depending on the authors. Sequences of both species (Arion ater and A. rufus) together with A. vulgaris and two outgroups (A. iratii and A. subfuscus) were downloaded from GenBank (https://www.ncbi.nlm.nih.gov/genbank/, last access: 12 June 2018) and aligned with the programme AliView v.1.17.1 (Larsson, 2014) in a different data set for each gene. In total, 68 DNA sequences were analysed, with more than 20 sequences per gene (Table 2).
Table 1Sample details of the employed data set including three mitochondrial genes (16s, COI and ND1) and five species Arion ater, A. rufus, A. vulgaris, A. iratii and A. subfuscus with their GenBank accession numbers.
Genetic variability of the three genes was measured as the number of polymorphic sites, the number of haplotypes, and the haplotype and nucleotide diversity with DNAsp v.5 software (Librado and Rozas, 2009). The pairwise genetic distances (K2P distance) between groups (potential species Arion ater and A. rufus) were estimated with MEGA v.7 (Kumar et al., 2016).
Species delimitation was done online (http://mptp.h-its.org/#/tree, last access: 12 June 2018) with the multi-rate Poisson tree process (mPTP; Kapli et al., 2017), a Bayesian implementation of the PTP model for species delimitation. This software has been shown to be the most accurate method for genetic species delimitations (better than previous PTP and popular distance-based methods), especially with the information on only one gene (Kapli et al., 2017). As mPTP is a method for species delimitation based on rooted phylogenetic trees, a phylogenetic input tree was previously done for each gene with BEAST v.2.4.5 (Bouckaert et al., 2014). Settings for the latter included the HKY substitution model, random local clock and a birth–death model under 20 million Markov chain Monte Carlo steps.
The genetic diversity of the three mitochondrial genes in each data set (359–544 base pair length) can be shown in Table 2. The number of haplotypes was 10 in the genes COI, 14 in the ND1 and 18 in the 16S. The haplotype diversity ranged between 0.848 (COI gene) and 0.981 (16S gene), and the nucleotide diversity ranged between 0.0774 (16S gene) and 0.1158 (ND1 gene). The K2P genetic distances between A. ater and A. rufus ranged between 9.16 % (16S gene) and 16.42 % (ND1 gene), all of which were much greater than the previously suggested 3 % of Hebert et al. (2004) for species differentiation. This result clearly points towards the classification of A. ater and A. rufus as different species. Because the genetic threshold for species identification is highly variable, there were discussions on which boundary is appropriate for different clades. For instance, even distances of 11 % have been found to signal different bird species (Tavares and Baker, 2008) but distances of less than 2 % distinguish water mites (García-Jiménez et al., 2017). Nevertheless, our results are far beyond the proposed 1 % divergence for species identification within this genus in the USA (Barr et al., 2009). However, based on this classical criterion (genetic distance) alone and a conservative stance, we considered that it was not enough to reach a conclusive answer (maybe this species is more recent, so their genetic distance is smaller), and more analyses were needed to solve the complex A. ater–A. rufus dilemma.
Table 2Genetic variability of the three amplified genes in the total data set, measured as the number of polymorphic sites (S), the number of haplotypes (H), the haplotype diversity (Hd) and the nucleotide diversity (Nd). n is sample size, length is sequence length in base pairs, K2P is pairwise genetic distances between A. ater and A. rufus sequences.
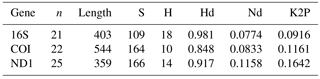
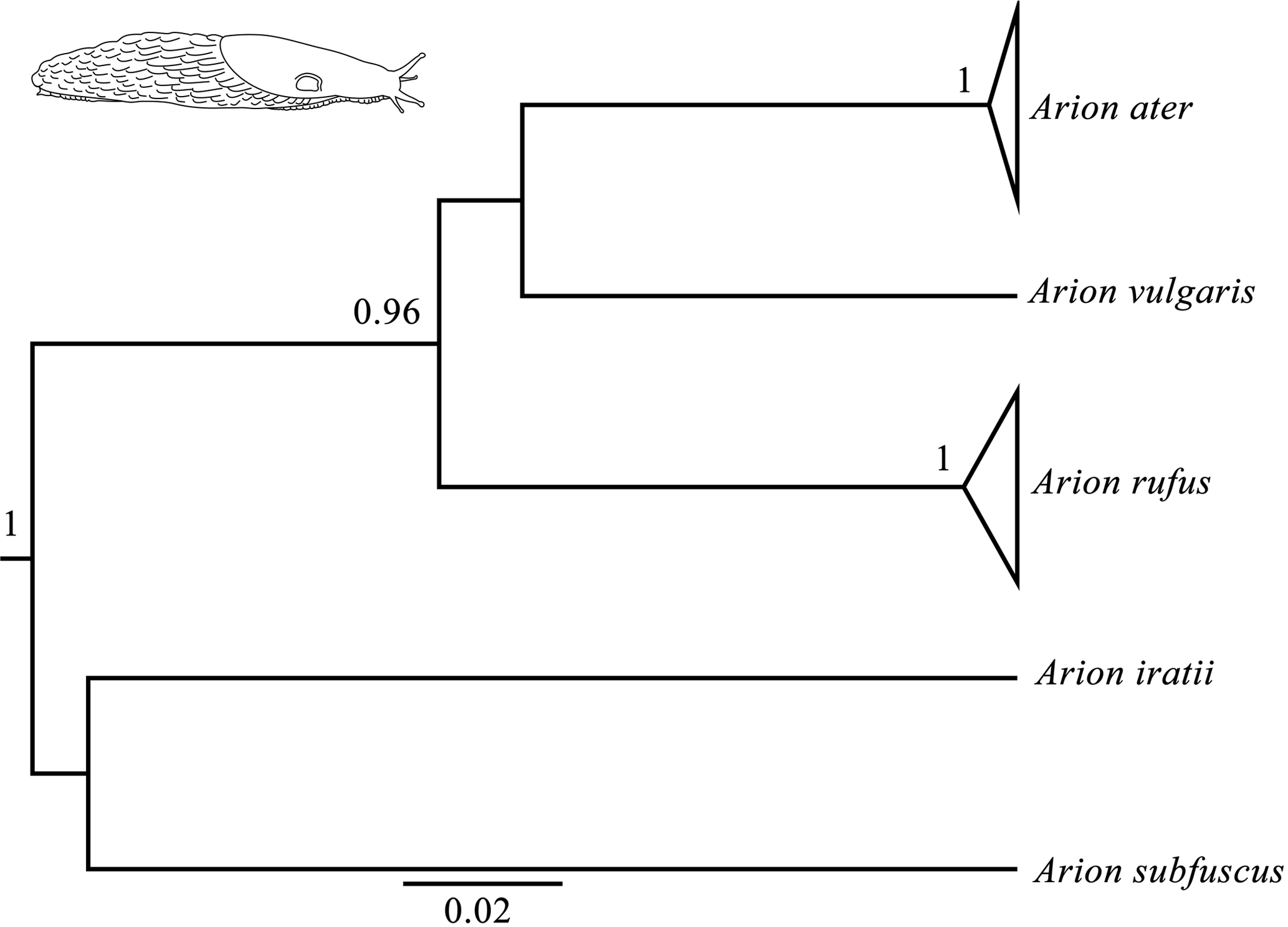
Figure 1Bayesian phylogenetic tree done with the COI gene (the same topology was found also found for the 16S and ND1 genes). Node numbers show Bayesian posterior probabilities higher than 0.96. Slug drawn by Daniel Martinez.
Bayesian phylogenetic trees were carried out independently for each of the three genes (16s, COI and ND1) and consistently showed high posterior probabilities in all cases (several of them higher than 0.96). All three phylogenetic trees were identical in their main topology (Fig. 1), showing three genetic lineages in addition to the outgroups (A. iratii and A. subfuscus): one with A. vulgaris, another comprising the sequences of A. ater and the last one including the sequences of A. rufus. The results obtained from these analyses confirm previous results, suggesting that both A. ater and A. rufus are independent evolutionary clades (Pfenninger et al., 2014; Rowson et al., 2014; Zemanova et al., 2016). Moreover, the large genetic distances between them (Table 2) also imply that these independent clades are not very recent, strongly supporting the idea that both clades identify different species and not subspecies or morphotypes, and confirm that morphological variability within each of the species (in all cases specimens were taken from several different distant geographical areas) is actually reflecting the existence of such subspecies and morphotypes.
In the case of mPTP analyses, which infer putative species boundaries from phylogenetic information (Zhang et al., 2013), we have also found highly congruent results independently of which gene was used. These results finally included three different species in the data set (in addition to the outgroups): Arion ater, A. rufus and A. vulgaris. It is important to mention that, although heteroplasmy has been described in this complex (Roth et al., 2012), it does not seem to affect our results in the sense that all the three independent genes from different individuals (and authors) have yielded the same results in all analyses. Putative hybrids would not be detected here because mitochondrial DNA is only maternally inherited, but the fact that all specimens of species grouped among them suggest that no hybrids are included in the data set, or that hybrids morphology is determined by mitochondrial DNA. mPTP analyses also agree, therefore, with genetic and phylogenetic results when distinguishing A. ater and A. rufus as different animal species.
This work therefore validates Arion ater and A. rufus as separate species for the first time, with three different mitochondrial genes and two different approaches. In this way our results shed light on discussions about these taxa being considered different morphological forms (Quinteiro et al., 2005), subspecies (Evans, 1986) or species (Bank et al., 2007) because of their very similar morphological characteristics. Our results also highlight the usefulness of the mPTP species delimitation software in difficult cases such as the A. ater–A. rufus complex, confirming in this case previous phylogenetic results, which are not enough for species delimitation on their own. On this basis, as in the case of cryptic species, the search of additional morphological correlates will allow proper identification of slug species in the field. As a final remark, Zemanova et al. (2016) pointed out that having obtained several hundred specimens from central and southern Iberia, none were A. ater or A. rufus. We have included in this work a sample of A. ater from Leon, Spain (GenBank accession number AY316229) and another from Portugal (GenBank accession number AY316228). These sequences were identified as A. ater by experts (Quinteiro et al., 2005) and phylogenetically grouped here with all the other A. ater from northern areas, confirming they are the same species.
Information regarding DNA sequences is in the material and methods section and in Table 1. All sequences have been downloaded from GenBank (https://www.ncbi.nlm.nih.gov/genbank, last access: 12 June 2018), a public and free genetic database.
DM and JLH designed the study, MLP and JLH analysed the data and all authors wrote and revised the manuscript.
The authors declare that they have no conflict of interest.
Jose L. Horreo was supported by a Spanish MINECO Juan de la
Cierva-Incorporación grant IJCI-2015-23618.
Edited by: Adrian Brennan
Reviewed by: Ted Von Proschwitz and two anonymous referees
Agrens, D., Fujisawa, T., Krammer, H.-J., Eberle, J., Fabrizi, S., and Vogler, A. P.: Rarity and incomplete sampling in DNA-based species delimitation, Syst. Biol., 65, 478–494, 2016.
Amstrong, K. F. and Ball, S. L.: DNA barcodes for biosecurity: invasive species identification, Philos. T. Roy. Soc. B, 360, 1813–1823, 2005.
Bank, R. A., Falkner, G., and Proschwitz, T.: A revised checklist of the non-marine Mollusca of Britain and Ireland, Heldia, 5, 41–72, 2007.
Barr, N. B., Cook, A., Elder, P., Molongoski, J., Prasher, D., and Robinson, D. G.: Application of a DNA barcode using the 16S rRNA gene to diagnose pest Arion species in the USA, J. Mollus. Stud., 75, 187–191, 2009.
Baum, D. A. and Shaw, K. L.: Genealogical perspectives on the species problem. In: Experimental and Molecular Approaches to Plant Biosystematics. in: Monographs in Systematic Botany from the Missouri Botanical Garden, edited by: Hoch, P. C. and Stephenson, A. G., 1995.
Blattmann, T., Boch, S., Türke, M., and Knop, E.: Gastropod seed dispersal: an invasive slug destroys far more seeds in its gut than native gastropods, PLos One, 8, e75243, https://doi.org/10.1371/journal.pone.0075243, 2013.
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., Suchard, M. A., Rambaut, A., and Drummond, A. J.: BEAST 2: A Software Platform for Bayesian Evolutionary Analysis, PLoS Comput. Biol., 10, e1003537, https://doi.org/10.1371/journal.pcbi.1003537, 2014.
Burnet, B.: Enzyme protein polymorphism in the slug Arion ater, Genet. Res., 20, 161–173, 1972.
Evans, N. J.: An investigation of the status of the terrestrial slugs Arion ater ater (l.) and Arion ater rufus (l.) (Mollusca, Gastropoda, Pulmonata) in Britain, Zool. Scr., 15, 313–322, 1986.
García-Jiménez, R., Horreo, J. L., and Valdecasas, A. G.: Minimal barcode distance between two water mite species from Madeira Island: a cautionary tale, Exp. Appl. Acarol., 72, 133–143, https://doi.org/10.1007/s10493-017-0147-5, 2017. 2017.
Gismervik, K., Bruheim, T., Rørvik, L. M., Haukeland, S., and Skaar, I.: Invasive slug populations (Arion vulgaris) as potential vectors for Clostridium botulinum, Acta Vet. Scand., 56, 65, https://doi.org/10.1186/s13028-014-0065-z, 2014.
Goldstein, P. Z. and De Salle, R.: Integrating DNA barcode data and taxonomic practice: determination, discovery and description, BioEssays, 33, 135–147, 2011.
Hagnell, J., Schander, C., and Proschwitz, T.: Hybridisation in arionids: the rise of a super slug?, in: British Council for Crop Protection symposium Proceedings, edited by: Dussart, D. B. J., 2003.
Hatteland, B. A., Solhoy, T., Schander, C., Skage, M., von Proschwitz, T., and Noble, L. R.: Introgression and differentiation of the invasive slug Arion vulgaris from native A. ater, Malacologia, 58, 303–321, 2015.
Hebert, P. D., Cywinska, A., Ball, S. L., and deWaard, J. R.: Biological identifications through DNA barcodes, P. Natl. Acad. Sci. USA, 270, 313–321, 2003.
Hebert, P. D. N., Stoeckle, M. Y., Zemlak, T. S., and Francis, C. M.: Identification of Birds through DNA Barcodes, Plos Biol., 2, https://doi.org/10.1371/journal.pbio.0020312, 2004.
Horreo, J. L.: “Representative Genes”, is it OK to use a small amount of data to obtain a phylogeny that is at least close to the true tree?, J. Evolution Biol., 25, 2661–2664, 2012.
Kapli, P., Lutteropp, S., Zhang, J., Kobert, K., Pavlidis, P., Stamatakis, A., and Flouri, T.: Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo, Bioinformatics, 33, 1630–1638, 2017.
Kumar, S., Stecher, G., and Tamura, K.: MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets, Mol. Biol. Evol., 33, 1870–1874, https://doi.org/10.1093/molbev/msw054, 2016.
Larsson, A.: AliView: a fast and lightweight alignment viewer and editor for large data sets, Bioinformatics, 30, 3276–3278, 2014.
Librado, P. and Rozas, J.: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data, Bioinformatics, 25, 1451–1452, 2009.
Lim, G. S., Balke, M., and Meier, R.: Determining species boundaries in a world full of rarity: singletons, species delimitation methods, Syst. Biol., 61, 165–169, 2012.
Noble, L. R. and Jones, J. S.: A molecular and ecological investigation of the large arionid slugs of North-West Europe: the potential for new pest specie, in: The ecology of agricultural pests: biochemical approaches, edited by: Symondson, W. O. C. and Liddell, J. E., Chapman & Hall, London, 1996.
Pfenninger, M., A., Weigand, A., Bálint, M., and Klussmann-Kolb, A.: Misperceived invasion: the Lusitanian slug (Arion lusitanicus auct. Non-Mabille or Arion vulgaris Moquin-Tandon 1855) is native to central Europe, Evol. Appl., 7, 702–713, 2014.
Quinteiro, J., Rodríguez-Castro, J., Castillejo, J., Iglesias-Piñeiro, J., and Rey-Méndez, M.: Phylogeny of slug species of the genus Arion: evidence of monophyly of Iberian endemics and of the existence of relict species in Pyrenean refuges, J. Zool. Syst. Evol. Res., 43, 139–148, 2005.
Renner, S. S.: A return to Linnaeus's focus on diagnosis, not description: the use of DNA characters in the formal naming of species, Syst. Biol., 65, 1085–1095, 2016.
Roth, S., Hatteland, B. A., and Solhoy, T.: Some notes on reproductive biology and mating behaviour of Arion vulgaris Moquin-Tandon 1855 in Norway including a mating experiment with a hybrid of Arion rufus (Linnaeus 1758) x ater (Linnaeus 1758), J. Conchol., 41, 249–257, 2012.
Rowson, B., Anderson, R., Turner, J. A., and Symondson, W. O. C.: The slugs of Britain and Ireland: undetected and undescribed species increase a well-studied, economically important fauna by more than 20 %, Plos One, 9, e91907, https://doi.org/10.1371/journal.pone.0091907, 2014.
Tavares, E. S. and Baker, A. J.: Single mitochondrial gene barcodes reliably identify sister-species in diverse clades of birds, BMC Evol. Biol., 8, 81, https://doi.org/10.1186/1471-2148-8-81, 2008.
Vogler, A. P. and Monagan, N.: Recent advances in DNA taxonomy, J. Zool. Syst. Evol. Res., 45, 1–10, 2007.
Zemanova, M. A., Knop, E., and Heckel, G.: Phylogeographic past and invasive presence of Arion pest slugs in Europe, Mol. Ecol., 25, 5747–5764, https://doi.org/10.1111/mec.13860, 2016.
Zhang, C., Rannala, B., and Yang, Z.: Bayesian species delimitation can be robust to guide-tree inference errors, Syst. Biol., 63, 993–1004, 2014.
Zhang, J., Kapli, P., Pavlidis, P., and Stamatakis, A.: A general species delimitation method with applications to phylogenetic placements, Bioinformatics, 29, 2869–2876, 2013.